

Δικαίωμα στην ελπίδα για τα βρέφη που γεννιούνται με Νωτιαία Μυϊκή Ατροφία (SMA) και τις οικογένειές τους προσφέρουν νέες, επαναστατικές προσεγγίσεις για την αντιμετώπιση αυτής της σπάνιας και απειλητικής για τη ζωή γενετικής, νευρομυϊκής ασθένειας.
Αποτέλεσμα της έλλειψης του λειτουργικού γονιδίου SMN1, που είναι υπεύθυνο για την παραγωγή μιας καταλυτικής για τη λειτουργία και την επιβίωση των κινητικών νευρώνων πρωτεΐνης, η SMA οδηγεί προοδευτικά σε μυϊκή αδυναμία, παράλυση έως και θάνατο των ασθενών.
Πιο συγκεκριμένα, η έλλειψη της εν λόγω πρωτεΐνης έχει ως συνέπεια, αμέσως μετά τη γέννηση, τη μη αναστρέψιμη απώλεια των κινητικών νευρώνων που επηρεάζουν όλες τις μυϊκές λειτουργίες, από την κατάποση και τη βασική κίνηση μέχρι την αναπνοή!
Τύποι της SMA
Αυτό εξαρτάται από τη σοβαρότητα της νόσου. Υπάρχουν τρεις βασικοί τύποι της SMA:
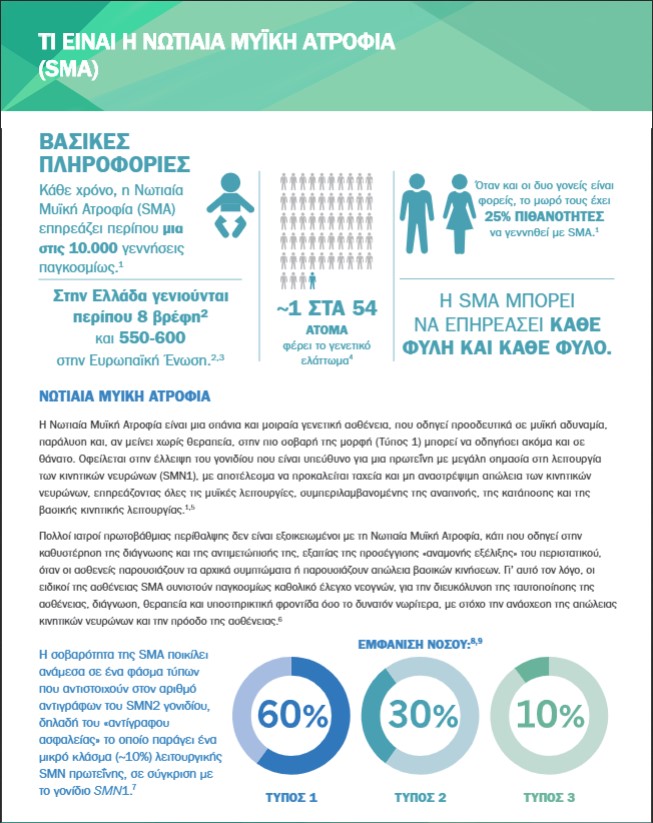
- SMA Τύπου 1: Αφορά στο 60% των περιπτώσεων και συνιστά την πιο σοβαρή μορφή της νόσου. Δίχως θεραπεία, οι ασθενείς έχουν απώλεια κίνησης εντός των πρώτων έξι μηνών από τη γέννηση, ενώ πριν συμπληρωθεί το δεύτερο έτος πάνω από το 90% των περιπτώσεων καταλήγουν ή χρειάζονται μόνιμη αναπνευστική υποστήριξη.
- SMA Τύπου 2: Αφορά στο 30% των ασθενών, οι οποίοι αδυνατούν να σταθούν χωρίς υποστήριξη και συνήθως χρειάζονται αναπηρικό καροτσάκι. Αν είναι σε θέση να καθίσουν χωρίς βοήθεια στα πρώτα παιδικά χρόνια, συχνά η ικανότητα αυτή χάνεται μετά τα μέσα της εφηβείας.
- SMA Τύπου 3:Αφορά στο 10% των περιπτώσεων και έχει ως βασικό χαρακτηριστικό τη δυσκολία στο τρέξιμο, το περπάτημα καθώς και το ανεβοκατέβασμα στις σκάλες. Οι ασθενείς ενίοτε χάνουν την ικανότητα να στέκονται ή να περπατούν χωρίς υποστήριξη με την πάροδο του χρόνου.
Εξάπλωση στον πληθυσμό
Η SMA εμφανίζεται μια φορά σε κάθε 10.000 γεννήσεις περίπου, σύμφωνα με διεθνείς επιστημονικές στατιστικές. Στην Ευρώπη, υπολογίζεται ότι κάθε χρόνο γεννιούνται περίπου 550-600 βρέφη με SMA, ενώ στην Ελλάδα οκτώ.
Αντίστοιχα, το γενετικό ελάττωμα εκτιμάται ότι το φέρουν περίπου 1 στα 54 άτομα. Η SMA έχει 25% πιθανότητα να εμφανιστεί όταν αμφότεροι οι γονείς είναι φορείς του γενετικού αυτού ελαττώματος.
Πώς αντιμετωπίζεται;
Απόσταγμα πολυετούς επιστημονικής έρευνας, σήμερα υπάρχουν δύο εγκεκριμένες θεραπευτικές επιλογές για την αντιμετώπιση της SMA:
- Φαρμακευτική αγωγή: έχει ως στόχο τη διαχείριση τω συμπτωμάτων. Χορηγείται σε επαναλαμβανόμενες δόσεις συντήρησης σε βάθος χρόνου, για το υπόλοιπο της ζωής του ασθενούς.
- Γονιδιακή θεραπεία: Πρόκειται για μια εφάπαξ θεραπεία, η οποία σχεδιάστηκε για να αντιμετωπίσει τη γενετική αιτία της νόσου. Εγκεκριμένη να κυκλοφορεί τόσο στις ΗΠΑ, όσο και στην Ευρώπη, η θεραπεία αυτή αντικαθιστά στα κύτταρα του ασθενούς τη λειτουργία του γονιδίου SMN1, που λείπει ή δεν λειτουργεί, με ένα νέο λειτουργικό αντίγραφο. Έτσι, σταματά η καταστροφή των κινητικών νευρώνων και αναστέλλεται η πρόοδος της νόσου.
Κλειδί η έγκαιρη διάγνωση
Είναι ευκταίο η διάγνωση της SMA να τίθεται έγκαιρα, ώστε να χορηγείται άμεσα η θεραπεία και να διασφαλίζεται η ελάχιστη δυνατή επίπτωση στους κινητικούς νευρώνες των ασθενών.
Αυτό στην πράξη δεν είναι εύκολο, όμως, καθώς τα πρώτα σημάδια της SMA είναι ανεπαίσθητα και διαφεύγουν της προσοχής πολλών γιατρών, που δεν έχουν γνώση της νόσου. Έτσι, τα παιδιά μπορεί να φαίνονται υγιή για αρκετό διάστημα μέχρι να γεννηθούν υποψίες και να τεθεί τελικά η διάγνωση, ενόσω εντωμεταξύ καταστρέφονται οι κινητικοί τους νευρώνες.
Για τον σκοπό αυτό, συστήνεται από πολλούς ειδικούς η ανάπτυξη ενός γενικευμένου προγράμματος νεογεννητικού ελέγχου για SMA, ώστε να διασφαλίζεται η έγκαιρη ανίχνευση της SMA αμέσως μετά την κύηση. Ήδη, σε αρκετές ευρωπαϊκές χώρες τρέχουν πιλοτικά προγράμματα αντίστοιχων νεογεννητικών ελέγχων.
Σημειώνεται πως σήμερα, έχει εγκριθεί η υπαγωγή όλων των σπάνιων ασθενειών για τις οποίες υπάρχει θεραπεία, όπως η SMA, σε έλεγχο νεογνών.
Αυτές οι πληροφορίες προορίζονται για γενική πληροφόρηση και ενημέρωση του κοινού και σε καμία περίπτωση δεν μπορούν να υποκαταστήσουν τη συμβουλή ιατρού ή άλλου αρμόδιου επαγγελματία υγείας



















{kind=link}